
学术资源
案例分享 | X连锁鱼鳞病
近日,我们收到了一位临床医生的咨询,他的患者是名2岁的男孩儿,临床表现为皮肤干燥,皮肤表层出现大面积的褐色鳞屑,身材矮小,语言发育落后等,临床怀疑可能为鱼鳞病。但由于鱼鳞病是一种具有遗传异质性的疾病,不同类型致病机制也会有所不同,从而导致医生无法抉择对该患儿采用哪种分子诊断方法。
什么是鱼鳞病呢?
鱼鳞病是一种以皮肤干燥、脱屑为主的遗传性皮肤病,可伴有系统损害而出现相应的综合征。按遗传机制、临床表现、组织病理的不同,大致可分为寻常型鱼鳞病、X-性连锁鱼鳞病(XLI)、板层状鱼鳞病、先天性非大疱性鱼鳞病样红皮病和火棉胶样婴儿等,其中以寻常型鱼鳞病和X性连锁鱼鳞病最为常见。
寻常型鱼鳞病(ichthyosis vulgaris,IV)
一种常染色体半显性遗传鱼鳞病,也是临床上最常见、症状最轻的一种鱼鳞病,发病率约为1/300。
IV属于半显性遗传,且受生活环境、空气湿度等各种条件的影响,因此个体间差异较大,临床症状也表现为多样性。IV患者的主要临床特点表现为儿童期开始发病,皮面出现细薄的片状鳞屑,同时伴有掌跖角化增厚,手臂、上臂、股外侧皮肤增厚等。而当生活在潮湿环境中时,IV患者可能没有任何临床症状。
注:半显性遗传,指当患者携带致病基因纯合子时,则其临床症状相对较严重;当患者携带致病基因杂合子时,则其临床症状相对较轻。
研究报道显示,IV的致病基因主要是定位于1q22上的FLG基因。FLG基因变异可导致半显性性状的发生,即杂合变异症状较轻或无症状显现,纯合变异或复合杂合变异则症状相对较重。
X性连锁鱼鳞病(X-linked ichthyosis,XLI)
一种X性连锁隐性遗传角化障碍性皮肤病,是鱼鳞病中最常见的一型。XLI主要发生于男性,大多数女性仅为携带者。男性发病率约为1/6000~1/2000。
XLI的临床表现以皮肤干燥及身体表面不同部位对称分布的大片状、多角形、深褐色鳞屑为主要特征。皮肤表现往往在夏季减轻,而冬季和干燥季节加重。此外,XLI同时伴有角膜混浊、隐睾症以及智力低下、身材矮小、神经系统异常等临床综合征。
IV和XLI虽均属于鱼鳞病,但其临床表现仍存在差异。
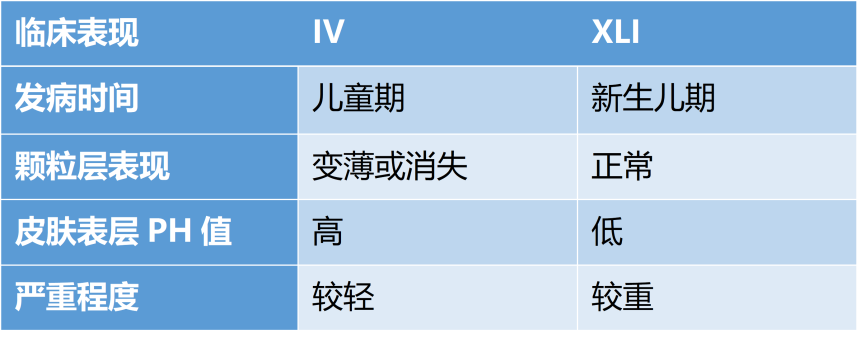
表1 IV与XLI临床表型的比较
研究证实,XLI的发病主要是由于STS基因变异所引起的。STS基因主要编码类固醇硫酸酯酶,一种广泛分布于哺乳动物组织中的微粒体酶,可以水解胆固醇硫酸盐。90%的XLI患者表现为STS全基因的缺失,少数表现为STS基因的部分缺失或点变异。STS基因的缺失引起角质层的类固醇硫酸酯酶缺失,导致表皮起屏障作用的胆固醇生成减少,从而导致角质层扁平细胞膜结构破坏,最终引起表皮屏障功能障碍。
鱼鳞病的诊断和治疗
鱼鳞病属于遗传性疾病,在临床上的诊断主要依赖于生化检测和基因诊断。基因诊断可通过高通量测序、定量PCR、Sanger测序、染色体芯片分析等方式进行针对性的遗传检测。目前,鱼鳞病无法彻底根治,治疗方式以缓解症状为主,增加角质层含水量和促进正常角化。
那对于该患儿采取什么检测方法呢
芯片检测结果
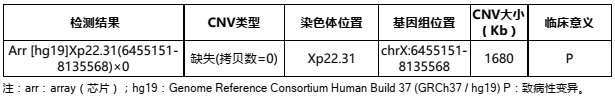
结果解读
检测结果显示,该患儿在Xp22.31区域存在约1.7Mb缺失。该区域仅包含一个OMIM致病基因STS,为X性连锁鱼鳞病的致病基因。根据ClinGen Dosage sensitivity数据库显示,该区域是已知的致病性区域,且在疾病数据库Decipher中记载有数十例病例有类似的缺失,这些病例临床表型包括:鱼鳞病、身材矮小、智力障碍等。
X性连锁鱼鳞病呈X连锁隐性遗传模式,当患者为男性时,其母亲多为携带者。故后续对患儿母亲也进行了染色体芯片分析,以验证该Xp22.31半合子缺失的变异来源。母亲的CMA结果显示携带有与患儿相同区域Xp22.31的杂合缺失(拷贝数为1),但母亲并无相关临床症状,表明母亲为鱼鳞病携带者。
综合上述结果,我们可知该患儿所携带的Xp22.31半合子缺失遗传自母亲,且是导致其鱼鳞病的主要原因。
通过本案例,我们可知充分了解疾病的致病机制有利于选择合适的检测方法,这不仅能够提高疾病的诊断率,帮助临床对疾病的诊疗,还能缩短患者确诊时间。